1.機能性表示食品の安全性評価
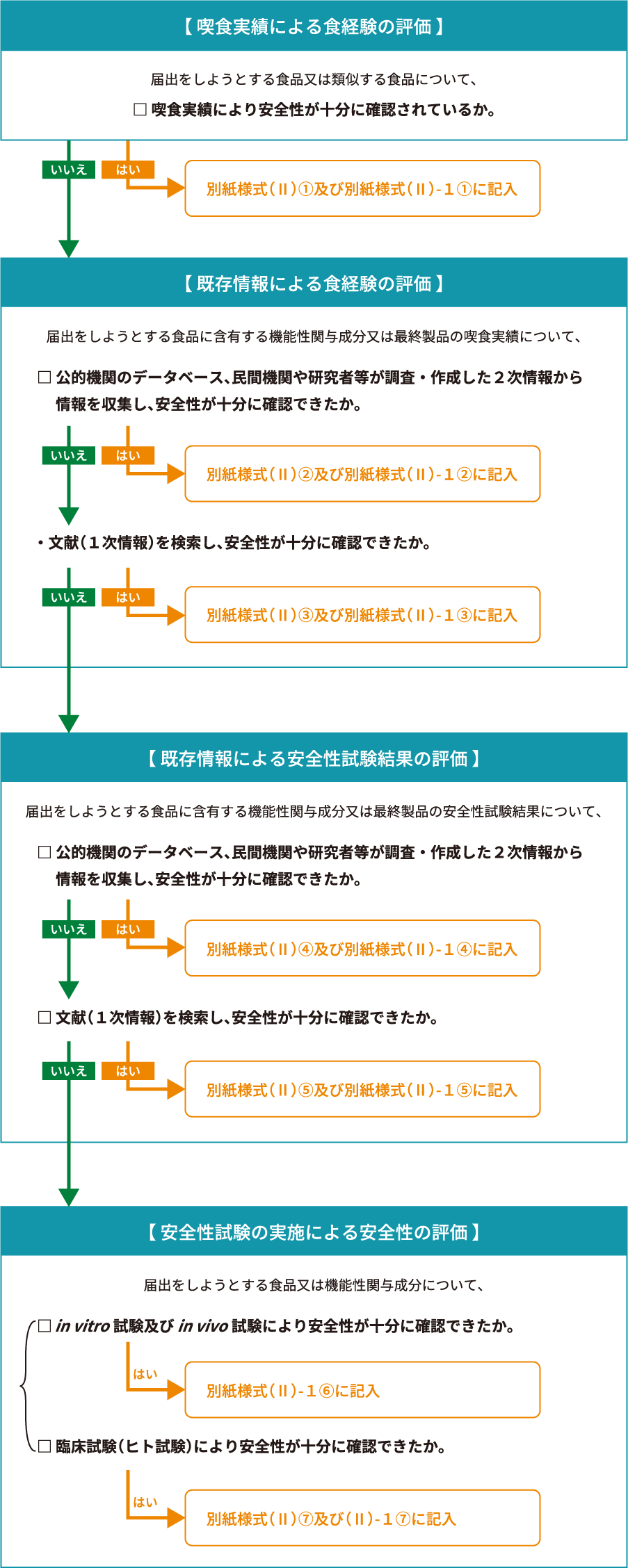
機能性表示食品の安全性の評価方法は大きく3つに分かれます
- 喫食実績による食経験の評価
- データベースの既存情報などを用いた情報収集
- 最終製品又は機能性関与成分における安全性試験の実施
安全性評価に関するフローチャート
- 参考:食品表示法の関係法令等
機能性関与成分の相互作用に関する評価を行い、相互作用がある場合は販売の適切性を説明できなければなりません。
- 機能性関与成分と医薬品の相互作用の有無を確認する
- 機能性関与成分を複数含む場合、当該成分同士の相互作用の有無を確認する
2.諸外国の食経験の考え方
十分な食経験の期間とは?
日本では、食品の十分とされる明確な食経験の期間は明確に定められていません。
しかし、アメリカ、ヨーロッパ、オーストラリア・ニュージーランドでは安全と考えられる食経験としての目安が明記されています。
-
- <アメリカ食品医薬品局(FDA)>
- 「食経験を構築するには、広範囲の使用が25年間あることが最低限」1)
-
- <欧州食品安全機関(EFSA)>
- 第三国の伝統食品をEUで認可承認する際の食経験として「その国の多くの人々により最低25年間、習慣的な食の中で使用された経験がある、又は継続的に使用されていること」2)
-
- <オーストラリア・ニュージーランド食品基準局(FSANZ)>
- 「2~3世代あれば使用歴として十分、5年以下では短いと考えられる、条件次第では10~20年でも十分な使用歴と考えられる」3)
- <参考文献>
-
- 1) U.S. Food and Drug Administration. Draft Guidance for Industry: New Dietary Ingredient Notifications and Related Issues. 2024.
- 2) European Food Safety Authority. Priority topics for the development of risk assessment guidance by EFSA’s Scientific Committee in 2016–2018. EFSA Journal. 2016;14(6).
- 3) Food Standards Australia New Zealand. FINAL ASSESSMENT REPORT PROPOSAL P291 REVIEW OF NOVEL FOOD STANDARD. 2007.
3.安全性試験の実施
ヒトを対象とした適正な安全性試験とは?
機能性表示食品制度における安全性試験は、「特定保健用食品の表示許可等について」(平成26年10月30日付け消食表第259号)を参照して、過剰摂取時及び長期摂取時における安全性を確認するための試験を実施することとなっています。
-
- 過剰摂取試験
- 一日摂取目安量の3倍量(一度に過剰量を摂取することが容易であると一般的に考えられる食品(食品形態が錠剤、カプセルなど)の場合には、原則として申請食品を用いて、一日摂取目安量の5倍量)
-
- 長期摂取試験
- 12週間以上の摂取期間
しかし、症例数や試験デザインに関する規定はなく、各事業者に判断を委ねられています。
では、コンセンサスのある安全性評価のヒト臨床試験は、どのような設定が考えられるのでしょうか。
オルトメディコでは以下のように考えます。
症例数と観察期間について
食品の安全性をヒト臨床試験にて評価する場合、医薬品開発におけるガイドラインが参考となり、症例数については、ICH-E1(致命的でない疾患に対し長期間の投与が想定される新医薬品の治験段階において安全性を評価するために必要な症例数と投与期間 1))が利用できます。
ICH-E1によると、期間が6ヶ月以下のヒト臨床試験の場合、短期間のうちに発現する有害事象の発現率(例えば、3カ月間の累積発現率が約1%)が明らかにされることが期待されています。また、妥当な頻度(一般的には0.5〜5%程度)の遅発性の有害事象が観察できるとともに、より高頻度に発現した有害事象がその後の期間中に増加するのか、あるいは減少するのかを観察できるだけの十分な症例数が必要であり、通常300600例の対象症例数が適当であるとされています。一方で、期間が12ヶ月以下のヒト臨床試験の場合、100例の患者に対して最低1年間投与して得られた成績は、安全性データベースの一部として採用できると考えられます。そのようなデータを得るためには、治験薬を予定される臨床用量で少なくとも1年間投与するように適切に計画されたプロスペクティブな試験(前向き研究)を実施すべきとされています。1年間の投与期間中に何ら重篤な有害事象が認められない場合には、そのような有害事象の1年間の累積発現率は3%未満と考えてよいとされています。
対象者について
特定保健用食品のように用途の範囲が定められている製品では、当該製品が想定する対象者を、摂取対象者が限定されない製品にいたっては、主な購買層に該当する対象者を、安全性評価のヒト臨床試験における対象者に設定することが一つ考えられます。なお、幅広い年齢層を対象に安全性評価を行う場合、前述の主な摂取対象者の安全性評価試験の結果が得られた後に、または、設定する症例数のうち、少なくとも 100 例を当該製品が想定する対象者もしくは主な購買層に該当する対象者とし、また各年代と生物学的性別が組み入れられるよう考慮することもできると考えられます。
調査項目について
一般生化学の血液項目に加え、製品に含有される機能性関与成分から想定される有害事象の発現件数が有用と考えます。なお、対象者背景の情報として、年齢、生物学的性別、摂取期間、栄養摂取状況、出産回数、閉経有無など、有害事象が発現した場合の因果関係を考察するに必要と考えられる項目の設定も望まれます。

- <参考文献>
-
- 1) 厚生労働省 . 致命的でない疾患に対し長期間の投与が想定される新医薬品の治験段階において安全性を評価するために必要な症例数と投与期間平成7年5月24日付審第592号厚生省薬務局審査課長通知 ) [Internet]. 1995 [cited 2024 Jan 4]. Available from: https://www.pmda.go.jp/files/000156199.pdf
<補足> 安全性試験の症例数について考える
4.機能性表示食品の安全性を担保するために
生産・製造および品質管理体制
機能性表示食品の安全性を確保するためには生産・製造の過程にて適切な品質管理を行うことが重要です。
製品の製造に関わる施設では、以下のような認証に基づく適切な品質の管理が望まれます。
-
- FSSC 22000
- Food Safety System Certification 22000の略。ISO 22000 に、食品安全のための前提条件プログラムを詳細化した ISO/TS 22002-1 等を加えたシステムであり、グローバル企業により積極的に推進されている。
-
- GMP
- Good Manufacturing Practiceの略。原材料の受入れから製造、出荷まで全ての過程において、製品が「安全」に作られ、「一定の品質」が保たれるようにするための適正製造規範。サプリメント形状の加工食品については、厚生労働省が GMP ガイドライン等を示して自主的取組を推進している。今後、機能性の観点も含めた GMPの検討が期待される。
-
- HACCP
- Hazard Analysis and Critical Control Pointの略。原材料の受入れから最終製品までの工程ごとに、①微生物、化学物質、金属の混入等による潜在的な危害を予測(危害要因の分析)した上で、②危害の発生防止につながる特に重要な工程(重要管理点)を継続的に監視・記録する工程管理のシステム。コーデックス委員会により、HACCP システムとその適用のためのガイドラインが示されている。
-
- ISO 22000
- International Organization for Standardization(国際標準化機構)が策定した規格の一つ。食品安全マネジメントシステムの一つであり、フードチェーンのあらゆる組織に対する要求事項のこと。危害要因を分析した上で重要管理点を継続的に監視・記録する工程管理システムを HACCP から、品質マネジメントシステムの考え方をISO 9001 から取り入れた ISO 規格。飼料生産者、収穫者、農家、材料の製造業者、食品製造業者、小売業者、食品サービス業者、清掃・洗浄及び殺菌・消毒サービス業者、輸送・保管及び配送業者等、フードチェーンに直接的又は間接的に関わる全ての組織を適用範囲とする。
参考:食品表示法の関係法令等
健康被害情報の収集
事前に安全性を十分に確認していても、医薬品と異なる食品は摂取が限定されないことから、健康被害が発生した場合その被害が急速に拡大する恐れがあります。被害の拡大、およびその健康被害を未然に防ぐためにも届出者は健康被害の情報を収集し、行政機関への報告を行う体制を十分に整備する必要があります。また、新聞やウェブサイト等を通じた消費者への情報提供を行う体制の整備も求められています。
届出後の品質管理
本制度では、届出後の機能性関与成分やその他の成分の安全性に関する分析の定期実施とその資料の保管が定められておりますが、その結果を一般公開することについては定められていません。しかし、だれでも見ることができるウェブサイト等で公開するなどの方法をとり、消費者が届出者に問合せを行うことなく簡便に確認できるようより積極的な情報公開に努めるのも届出者の責任です。